Ученые раскрыли самый полный геном человека всех времен

Молекулярные биологи завершили секвенирование генома человека в специальном выпуске журнала Science. В предыдущей версии генома, появившейся в 2001 году, около 8 процентов последовательности оставались нерасшифрованными. В основном это некодирующие участки, а также центральные и концевые участки хромосом.
Полная версия генома позволяет более точно идентифицировать индивидуальные генетические особенности людей и может стать новым стандартом в генетике, несмотря на то, что в ней по-прежнему отсутствует целая хромосома.
Секвенирование ДНК человека: основы
В 2000 году Human Genome Project и компания Крейга Вентера Celera Genomics объявили о завершении секвенирования последовательности ДНК человека. К 2001 году они опубликовали свои черновые варианты сборки, а к 2003 году объединили свои усилия и наработки, чтобы собрать единую чистую копию.
Он стал первым эталоном, или эталонным геномом, по которому проверялись все, кто расшифровывал новые геномы человека или искал генетические причины болезней. Однако работа по чтению ДНК человека на этом не закончилась.
Первая версия генома человека не была полной
Авторы первой версии генома человека не скрывали, что он далеко не полный. Например, осталось 341 место. Кроме того, в своей работе исследователи опирались на эухроматин, ту фракцию ДНК, которая обычно находится в клетке в рыхлом состоянии и с которой можно считывать информацию.
Таким образом, первая версия генома не включала многих участков гетерохроматина, «скрученной» фракции ДНК. Он состоит в основном из последовательностей, не кодирующих белки, но выполняющих различные технические и структурные (и часто не до конца понятные) функции — следовательно, они также могут влиять на жизнь и работу клетки.
В первой версии генома тоже было не до конца понятно, какие гены и некодирующие участки за что отвечают. Это выясняется, например, проектом ENCODE.
Наконец, эталонный геном не в полной мере учитывал генетическое разнообразие людей — несмотря на то, что он был собран из случайных количеств ДНК от нескольких десятков человек. Для заполнения этих пробелов были предприняты другие проекты, такие как «Тысяча геномов».
С тех пор геном неоднократно уточнялся, появилось несколько обновленных ссылок. Последний, GRCh38.p13, был опубликован в 2019 году. Но даже в нем было много белых пятен — областей, где вместо нуклеотидов были указаны буквы N, или где были заменены какие-то суррогатные последовательности.
Ученые секвенировали полный геном человека
Консорциум «От теломеры к теломере» (Т2Т-Консорциум, теломера — терминальный участок хромосомы) взял на себя миссию по достройке недостающих частей генома человека.
В него вошли ученые из 54 институтов и лабораторий разных стран, а результатом их работы стала первая полноценная сборка генома — о которой они рассказали в шести статьях в журнале Science.
Все, что мы знаем о геноме CHM13
Первая статья представляет собой презентацию новой сборки, в которой авторы рассказывают об используемых ими методах и подводят итоги своей работы. Новый геном был назван CHM13 в честь культуры клеток, ставших донорами ДНК.
Эта культура происходит от пузырного заноса, необычной опухоли человека, которая появляется, если оплодотворенная яйцеклетка по какой-то причине теряет материнские хромосомы.
Пузырный занос удобен тем, что часто его геном состоит из удвоенного набора хромосом, принесшего с собой сперматозоид. Это означает, что обе копии каждой хромосомы должны быть практически идентичными (за исключением точечных мутаций и случайных разрывов), и при секвенировании не нужно выяснять, какая из копий расположена на том или ином участке.
Сборка CHM13 отличается от своих предшественников технологией секвенирования. Предыдущие версии генома собирались из множества коротких последовательностей — то есть ДНК сначала разбивали на небольшие участки, считывали каждый отдельно, а затем накладывали друг на друга.
Но этот метод не подходит для гетерохроматина, так как там много повторяющихся участков, в расположении и количестве которых легко ошибиться (например, некоторые гены рибосомной РНК у человека могут иметь 300-400 копий).
Поэтому участники T2T Consortium использовали метод длинных чтений (long-read sequencing), то есть разбивали ДНК на длинные части и считывали их целиком.
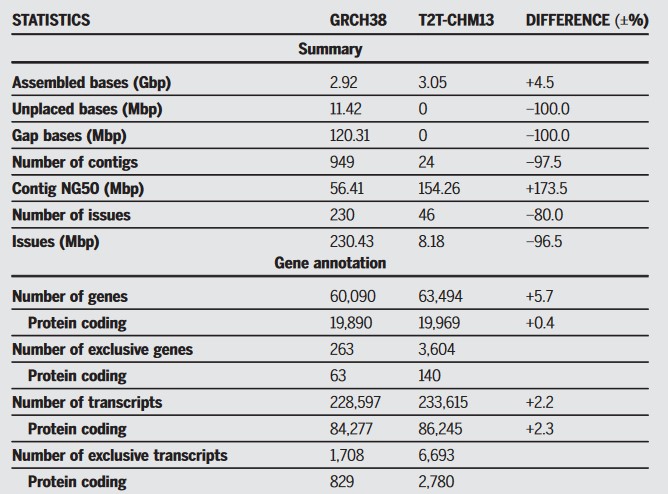
В результате CHM13 включал 3 054 815 472 п.н. ядерной ДНК и 16 569 п.н. митохондриальной ДНК. Из них 182 млн пар совершенно новые: их не было в предыдущей сборке генома 2019 года. В этом геноме, отмечают авторы работы, нет пробелов и нуклеотидов, которым не нашлось бы места — он полностью завершен.
Подавляющее большинство новых участков представляют собой некодирующие ДНК, в основном центромерные (то есть из середины хромосом, в том месте, где они скрепляются друг с другом характерным перекрестом при мейозе).
Однако исследователям удалось найти новые гены — всего 1956 штук. Из них около сотни, по их оценкам, кодируют белки (остальные могут кодировать отдельные типы РНК или вообще не работать).
Что включали остальные пять статей?
Остальные пять статей этого номера посвящены отдельным углубленным исследованиям в рамках проекта. Например, в одной из работ рассказывается о центромерах, их разнообразии, строении и эволюции.
В другом — о повторах в геноме: авторы искали среди них ретротранспозоны (мобильные генетические элементы, способные перемещаться по геному или вставлять в него новые копии самих себя), в том числе и активные.
Третий касается сегментарных дупликаций, длинных отрезков с небольшим количеством копий, которые, вероятно, сыграли роль в эволюции приматов. Четвертая представляет собой карту метилирования вновь секвенированных областей.
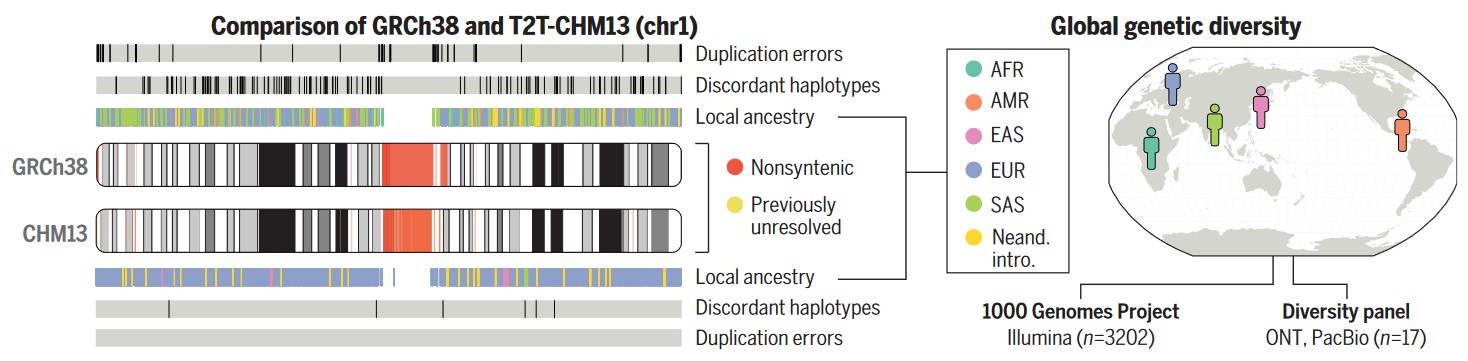
Наконец, еще одна статья посвящена практическому применению нового генома. Его авторы проверили, насколько удобно использовать сборку CHM13 для сравнения с ней отдельных геномов и поиска особых вариантов последовательностей.
Для этого они использовали базу данных проекта «Тысяча геномов» и, сравнивая последовательности из базы данных с CHM13, нашли более миллиона вариантов генов (тех, которые не были показаны при сравнении со сборкой GRCh38).
Поэтому члены консорциума предложили обозначить CHM13 как новый стандарт для генетических и геномных исследований.
Но и на этом расшифровка генома человека не закончится. У CHM13 есть свои недостатки — например, в этой сборке нет Y-хромосомы.
Это связано с тем, что клетки пузырного заноса несут по две идентичные копии каждой хромосомы, а генотип YY нежизнеспособен. Поэтому эту хромосому придется собирать отдельно.
Кроме того, CHM13 — это не синтетический геном из клеток разных людей, как это было с предыдущими сборками, а геном одной клеточной линии.
Поэтому Консорциуму придется собирать другие варианты геномов, чтобы их стандарт учитывал не только полную последовательность ДНК, но и разные ее варианты.
Поделитесь в вашей соцсети






